Visualización de secciones de la secuencia del genoma humano en el Instituto Wellcome Sanger cerca de Cambridge, Reino Unido.Crédito: Biblioteca de fotografías James King Holmes / Ciencia
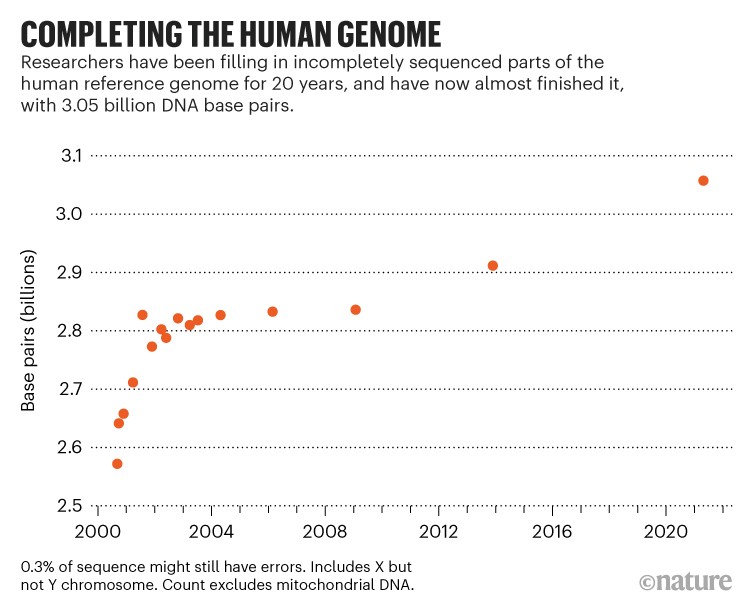
Cuando el genoma humano fue secuenciado hace dos décadas por Human Genome Project y la empresa de biotecnología Celera Genomics, la secuenciación no estaba realmente completa. Faltaba alrededor del 15%: las limitaciones tecnológicas han dejado a los investigadores incapaces de averiguar cómo encajan ciertos tramos de ADN, especialmente aquellos con muchas letras repetidas (o pares de bases). Científicos Resuelve algunos acertijos con el tiempo, pero el último genoma humano, que los genetistas han utilizado como referencia desde 2013, aún carece del 8% de la secuencia completa.
Ahora, los investigadores del Consorcio Telomere-to-Telomere (T2T), una colaboración internacional de unas 30 instituciones, han llenado esos vacíos. 27 de mayo antes de la impresión1 Titulada «La secuenciación completa del genoma humano», la investigadora de genómica Karen Megah de la Universidad de California en Santa Cruz y sus colegas informan que secuenciaron el resto, en el proceso de descubrir alrededor de 115 nuevos genes que codifican proteínas, para un total de 19,669 .
«Es emocionante tener algunas soluciones para áreas problemáticas», dice Kim Pruitt, experto en bioinformática del Centro Nacional de Información Biotecnológica de EE. UU. En Bethesda, Maryland, y describe el resultado como un «hito importante».
Nueva tecnología de secuenciación
El genoma recién secuenciado, apodado T2T-CHM13, agrega casi 200 millones de pares de bases a la edición 2013 de la secuencia del genoma humano.
Esta vez, en lugar de tomar ADN de una persona viva, los investigadores utilizaron una línea celular derivada de lo que se conoce como mola hidatiforme completa, un tipo de tejido que se forma en los humanos cuando un espermatozoide fertiliza un óvulo sin núcleo. La célula resultante contiene solo cromosomas del padre, por lo que los investigadores no tienen que distinguir entre dos juegos de cromosomas de diferentes personas.
Mega dice que la hazaña podría no haber sido posible sin una nueva tecnología de secuenciación de Pacific Biosciences en Menlo Park, California, que utiliza láseres para escanear largos tramos de ADN aislados de células, hasta 20.000 pares de bases a la vez. Los métodos de secuenciación convencionales leen el ADN en piezas de unos pocos cientos de pares de bases a la vez, y los investigadores vuelven a ensamblar estos tramos como si fueran piezas de un rompecabezas. Las piezas más grandes son mucho más fáciles de ensamblar, ya que es más probable que contengan secuencias superpuestas.
Sin embargo, T2T-CHM13 no es la última palabra en el genoma humano. El equipo de T2T tuvo problemas para resolver algunas regiones de los cromosomas y estimó que alrededor del 0,3% del genoma puede contener errores. No hay lagunas, pero Miga dice que las pruebas de control de calidad han resultado difíciles en esas áreas. Y el espermatozoide que formó la mola hidatiforme portaba el cromosoma X, por lo que los investigadores aún no han dispuesto el cromosoma Y, que generalmente conduce al desarrollo biológico masculino.
Cientos de genomas a seguir
T2T-CHM13 representa el genoma de una sola persona. Pero el T2T Consortium se ha asociado con un grupo llamado Human Pangenome Reference Consortium, que tiene como objetivo durante los próximos tres años secuenciar más de 300 genomas de personas en todo el mundo. Miga dice que los equipos podrán usar T2T-CHM13 como referencia para comprender qué partes del genoma tienden a diferir entre individuos. También planean secuenciar un genoma completo que contiene cromosomas de ambos padres, y el grupo Miga está trabajando en secuenciar el cromosoma Y, utilizando los mismos métodos nuevos para ayudar a llenar los vacíos.
Miga espera que los investigadores en genética descubran rápidamente si alguna de las regiones recientemente secuenciadas y los genes potenciales están asociados con enfermedades humanas. «Cuando salió el genoma humano, no teníamos las herramientas listas y listas para funcionar», dice, pero la información sobre la función de los genes recién secuenciados debería llegar más rápido ahora, porque «hemos construido muchos recursos.»
Ella espera que las futuras secuencias del genoma humano cubran todo, incluidas las secciones recién secuenciadas, no solo las porciones fáciles de leer. Esto debería ser más fácil ahora que se ha completado el genoma de referencia y se han resuelto algunos obstáculos técnicos. «Necesitamos alcanzar un nuevo estándar en genómica donde esto no sea algo especial, sino una rutina», dice.

«Erudito en viajes incurable. Pensador. Nerd zombi certificado. Pionero de la televisión extrema. Explorador general. Webaholic».